What is pancreatic cancer?
An overview of the disease
Pancreatic cancer occurs when a malignant tumor forms in the pancreas, a vital organ in the digestive system.
Globally, around 496,000 new cases are diagnosed each year, with over 104,000 in Europe.
In the UK, about 10,500 people are diagnosed annually. Pancreatic cancer affects men and women equally with incidence increasing from the age of 45. The average age at diagnosis is 75.
The pancreas
The pancreas, about six inches long, lies behind the stomach and in front of the spine.
It has two main functions in the body:
- Enzymes: It produces enzymes that help digest food. These enzymes travel through the pancreatic duct to the small intestine.
- Hormones: It produces hormones like insulin and glucagon, which regulate blood sugar levels.
The pancreas has two types of glands:
- Exocrine Glands: Produce digestive enzymes.
- Endocrine Glands: Produce hormones that control blood sugar.
A tumor in the pancreas can block the bile duct, leading to jaundice (yellowing of the skin and eyes). Understanding these basics helps in grasping the impact of pancreatic cancer.
Understanding cancer
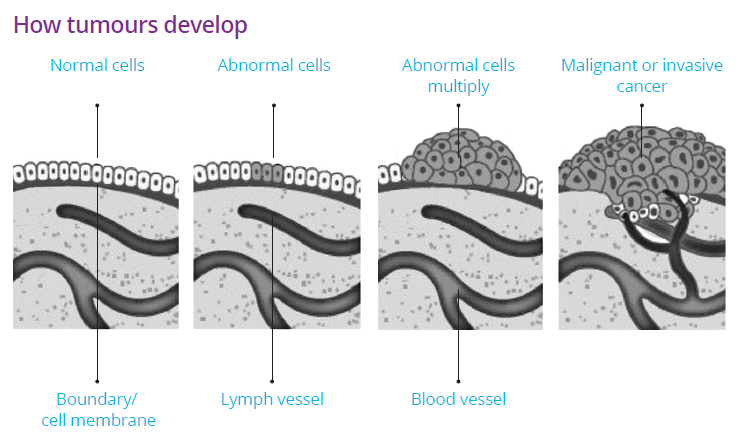
Cancer starts when cell DNA is damaged, leading to uncontrolled growth. Normally, cells grow, divide, and die in a regulated manner. When this process is disrupted, cells may form a tumor, which can be benign (non-cancerous) or malignant (cancerous). Malignant tumors can spread to other parts of the body, a process known as metastasis.
The two main types of pancreatic cancer
There are two main types of pancreatic cancer. Tumours that affect the exocrine glands, and ones that affect the endocrine glands.
-
Exocrine tumours make up most of all pancreatic cancers.
The most common type of exocrine tumour is called pancreatic ductal adenocarcinoma (PDAC) and comes from cells that line the ducts in pancreas that carry digestive juices into the intestine. There are other rarer kinds of exocrine tumours of the pancreas including Aden squamous carcinomas and undifferentiated carcinomas.
-
Neuroendocrine tumours (NETs) can be either benign (non-cancerous) or malignant (cancerous) and develop in the hormone-producing cells of the neuroendocrine system, which is spread throughout the body. NETs can appear in different organs, including the pancreas.
Pancreatic neuroendocrine tumours (pNETs) form in the pancreas' hormone-producing cells. These are less common than other pancreatic tumours and are classified as either functional (hormone-producing) or non-functional (non-hormone-producing). Functional tumours are usually detected earlier due to symptoms caused by hormone overproduction. Non-functional tumours don’t produce hormones but are more likely to be cancerous.
Most NETs occur randomly, but some people have a genetic condition, such as Multiple Endocrine Neoplasia Type 1, that increases their risk.
Types of pNETs include:
- Insulinoma: Produces too much insulin, causing symptoms like low blood sugar, frequent urination, and headaches.
- Gastrinoma: Produces gastrin, leading to stomach ulcers, cramps, and greasy stools.
- Somatostatinoma: Affects digestion and blood sugar control, causing diarrhoea, weight loss, and diabetes symptoms.
- VIPoma: Causes loose stools, dehydration, and fatigue.
- Glucagonoma: Raises blood sugar, leading to diabetes symptoms, skin rashes, and mouth sores.
Functional tumours cause symptoms due to hormone overproduction, while non-functional tumours are more likely to be cancerous but may not show symptoms until later.
What is Pancreatic cancer and how is it diagnosed?
This booklet for patients and carers describes pancreatic cancer, its causes and symptoms. It gives detailed information on the diagnostic tests used and the stages of pancreatic cancer. It includes a section on what to ask your doctor, where to go for further information and a glossary to explain many of the terms used.
Read more
The information provided in this site, or through links to other websites, is not a substitute for medical or professional care and should not be relied upon as such. Read our disclaimer.
Sources and references for this information product will be supplied on request. Please contact us quoting the Information Product number below:

- Information Product No: PCA0011v3
- Published: 26 Sep 2022
- Last updated: 26 Sep 2022
- Next Review Due: 3 Oct 2025